


Angiomatoid Fibrous Histiocytoma
Synopsis of VIVA-Asia Rare Tumour Board Webinar
10 January 2023 | 8:00PM - 9:00PM
Introduction
Angiomatoid fibrous histiocytoma (AFH) is a rare, slow-growing soft tissue tumor with intermediate biologic potential, primarily affecting children and young adults. It often presents as a superficial, painless lesion but can occur in deep or unusual locations, complicating diagnosis. Advances in molecular pathology, particularly the identification of gene fusions like EWSR1/CREB1, have improved diagnostic accuracy. This synopsis summarizes the regional experience and a review of literature discussed during the VIVA Rare Tumor Board on Jan 10, 2023.
Case Study 1 (Dr Chunyu Li, HKU-Shenzhen Hospital, China)
A 12-year-old Chinese boy presented with right axillary pain in October 2021. The pain worsened when he was writing, and no definitive treatment was provided despite multiple medical consultations. A year later, he noticed a bump in his right axilla. A preoperative MRI of the primary tumor, using axial non-contrast T1-weighted and fat-suppressed T2-weighted imaging, revealed a 3 cm lobulated mass in the right axilla. The mass showed a slightly hyperintense signal on T1-weighted imaging and an obviously hyperintense signal on T2-weighted imaging. Additionally, multiple lymph nodes were observed in the right axilla. A needle biopsy suggested a benign tumor, and he underwent surgery at a local hospital. Unfortunately, the tumor margin was positive, and the axillary lymph nodes were not explored.
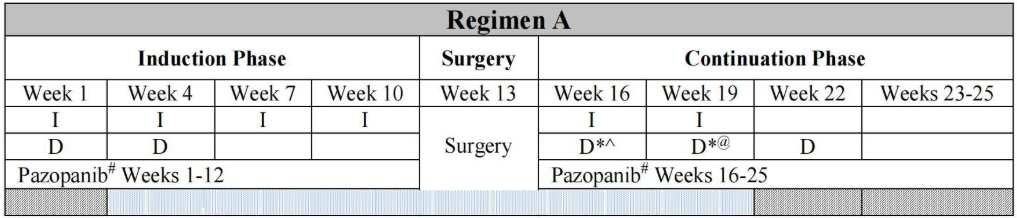
The pathology report initially indicated synovial sarcoma, prompting his transfer to HKU-Shenzhen Hospital for further evaluation. Upon further review by the pathologist, the tumor was found to have a pseudo-fibrous capsule with brownish hemosiderin deposits. A pericapsular rim of lymphoid cells with germinal centers was also noted. Cystic, aberrant blood vessel-like structures formed by tumor cells, rather than endothelial cells, were observed. The tumor cells were epithelioid or spindle-shaped, with some cells exhibiting a typical vesicular nucleus resembling histiocytes. Mitotic figures were scanty. Immunohistochemistry results were positive for H3K27me3, p16, Desmin, CD-99, and TLE-1, but negative for S100, SMA, SOX-10, Myogenin, MyoD1, and STAT-6. The Ki67 index was only 10%. These findings were consistent with angiomatoid fibrous histiocytoma (AFH) rather than synovial sarcoma. Fluorescence in situ hybridization (FISH) identified an EWSR-1 gene rearrangement. Subsequent tumor methylation profiling did not cluster with any known reference, and next-generation sequencing (NGS) revealed an EWSR1/CREB1 fusion. Staging workup, including CT, MRI, and bone scans, showed no evidence of metastases. The patient was treated according to the non-rhabdomyosarcoma soft tissue sarcoma regimen (ARST1321, Table 1). After two cycles of chemotherapy combined with pazopanib, a PET-CT scan detected a hypermetabolic nodule in the liver (SUVmax 3.3). MRI also revealed multiple enhancing nodules in the liver. However, a liver biopsy found no evidence of malignancy, and the nodules disappeared three weeks later. The patient completed his chemotherapy and has remained well for over 25 months since then.
Table 1. Children’s Oncology Group ARST1321 Treatment Regimen
Case Study 2 & Review of Literature (Dr Debbra Chong, KK Women & Children's Hospital, Singapore)
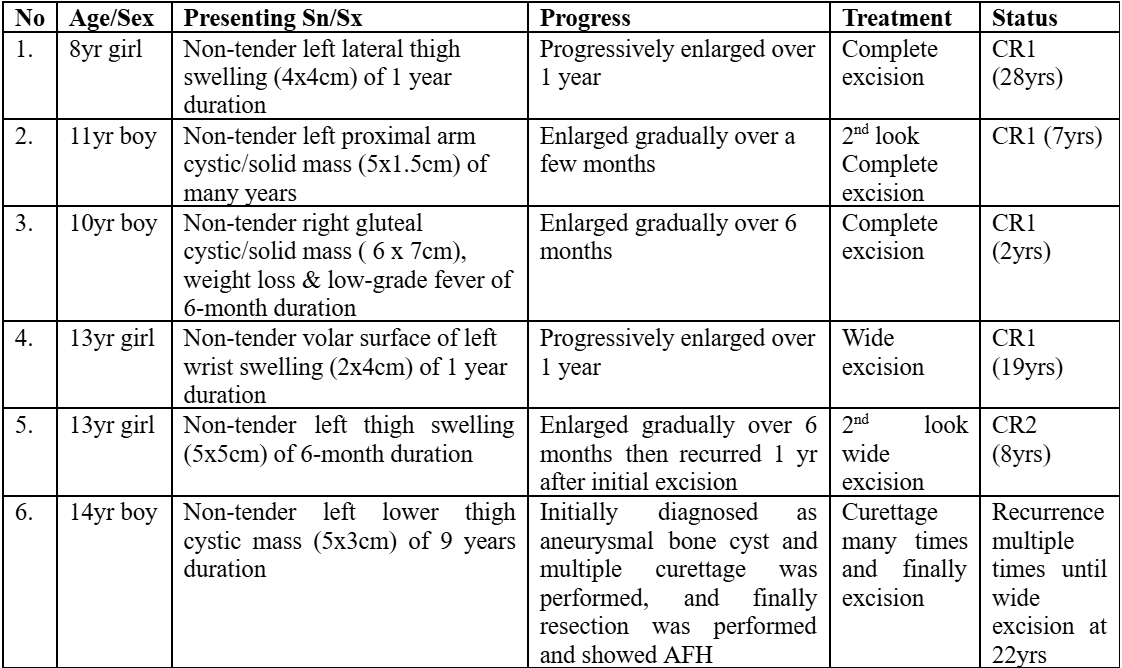
An 11-year-old girl that was transferred from an outside hospital for on and off fever of 20 days duration. She also had dizziness, headaches, intermittent abdominal pain, and 4 episodes of syncope. No other constitutional symptoms were mentioned. Her full blood count showed just mild anemia (Hb 10.2g/dL). The total white cells and platelets were normal. She had significantly elevated CRP (111mg/L) and ESR (120 mm/hr.). Ferritin was also elevated (504µg/L). Her urine HVA and VMA were not elevated, and her bone marrow morphology was unremarkable. Other than mildly raised ALT and AST, her renal and liver functions were normal. An extensive autoimmune workup was negative. Extensive viral, bacterial, fungal investigations were all negative. The CXR and ultrasound abdomen, pelvis was normal. She was given a broad spectrum of antibiotics, but her fever did not settle, PET-CT was finally performed and showed a hypermetabolic left adrenal mass 2.5x2.8x2.5 cm. An endoscopic left adrenalectomy was then performed. There was a 3cm encapsulated tumor in the superior medial aspect of the upper core of the left kidney. And the tumor was surrounded by fat and the normal adrenal gland, but the left kidney was otherwise grossly normal. Right after the surgery, her fever settled promptly. Pathology came back as an adrenal AFH with an EWSR1/ATF1 gene fusion. Currently, she is >5 years post-surgical removal of the tumor with no recurrence. Table 2 summaries the KK Children’s Hospital experience with such rare diagnosis.
Table 2. Case series of AFH at KK Children’s Hospital, with an additional case from Hong Kong
Review from the literature indicated mostly case reports or small series with up to 7 patients only. AFH is a rare, slow growing soft tissue tumor with an intermediate biologic potential. It is from an unknown line of differentiation and mostly involves children and young adults. In most patients, this lesion is present as a superficial, painless, slowly growing, and fluctuant, cutaneous lesion, usually on the extremities. Rarely, it can occur in unusual deep locations such as the female genital tract, bone, brain, the retroperitoneum, mediastinum, and visceral organs. Some patients may present with an associated paraneoplastic syndrome related to cytokine production. This can manifest in one of three ways, anemia, weight loss, or fever. In these cases, they usually have elevated inflammatory markers including CRP and ESR. Their IL6 level is also high. Interestingly, after complete excision, all these signs and symptoms will subside. Wide local resection is important because the margin status is a very crucial prognostic factor. Generally, recurrence risk is about 15%. The risk of metastasis is as low as 1%, but generally less than 5% is quoted in the literature.
Imaging features of AFH can be quite nonspecific. X rays are generally not useful. Contrasted CT may show a heterogeneous mass with cystic and enhancing components. Ultrasounds may show cystic features. For MRIs, generally, it shows homogeneously in isointensity T1 signals and heterogeneous hyperintense foci to T2 weighted imaging. Gadolinium enhancement may show a variegated internal and nodular appearance. And some cases may have a double rim sign as well, which consists of a rim of high signal intensity and adjacent rim of low signal intensity. But this does not occur in all patients. So, the MRI findings can be nonspecific, and the initial diagnosis of most patients were often wrong. Diagnosis and treatment are mainly by surgical excision.
Pathology of AFH (Dr Kenneth Chang, KK Women & Children's Hospital, Singapore)
Most cases of AFH exhibit a hemorrhagic cystic appearance. There are four characteristic histological features that aid in the diagnosis of AFH:
1. Lymphoid Cuff: The classic lesion is surrounded by a lymph node-like cuff, which may occasionally contain germinal centers.
2. Fibrous Capsule: The lesion typically has a thick fibrous capsule.
3. Blood-Filled Channels: These are blood-filled spaces that contribute to the hemorrhagic appearance of the lesion.
4. Histiocytoid Cells: The presence of numerous histiocytoid cells is a key feature.
These four histological features collectively enable pathologists to make a confident diagnosis of AFH. However, challenges may arise when the diagnosis is based on a Trucut biopsy, as the specimen obtained may vary depending on the needle's position. In such cases, the sample may resemble a benign lymph node, fibrosis, venous malformation, or even a histiocytic lesion. Therefore, it is essential to evaluate the full complement of all four characteristic histological features to ensure an accurate diagnosis.
In some instances, patients may not exhibit all four histological features, making the diagnosis more challenging. In such cases, immunophenotyping can provide additional diagnostic support. While the immunophenotype of AFH is variable, certain markers are characteristically positive, including SOX9 and Desmin. Other markers, such as SMA, EMA, and histiocytic markers like CD68 and CD163, may show variable expression. Importantly, certain markers should be negative to rule out differential diagnoses, such as rhabdomyosarcoma and other malignant tumors. Specifically, MyoD1, Myogenin, and cytokeratin should be negative in AFH lesions.
Advances in molecular pathology have significantly enhanced the diagnostic process for AFH. The majority of AFH cases are characterized by a specific gene fusion, EWSR1/CREB1. A smaller proportion of cases involve an alternative gene partner, ATF1. More recently, rare gene fusions, such as FUS/ATF1 and EWSR1/CREM1, have been reported in case studies. While the two most common fusions, EWSR1/CREB1 and EWSR1/ATF1, are also observed in clear cell sarcoma of soft parts, the distinction between AFH and clear cell sarcoma can be made based on morphological features. Clear cell sarcoma of soft parts exhibits downstream activation of the microphthalmia transcription factor pathway, resulting in positive melanocytic markers, which are typically negative in AFH. Additionally, recent case reports have described unusual intracranial mesenchymal tumors and malignant epithelioid tumors of the peritoneal cavity that share these gene fusions.
To identify specific molecular alterations, several techniques are available:
- Fluorescence In Situ Hybridization (FISH): This method is sensitive for detecting EWSR1 or FUS translocations. However, it does not identify the fusion gene partner, which may limit its utility in some cases. If the clinical and histological features strongly suggest AFH, demonstrating an EWSR1 translocation via FISH may suffice for diagnosis.
- Reverse Transcription Polymerase Chain Reaction (RT-PCR): While RT-PCR was initially used to identify gene fusions, its practicality is limited due to the highly variable breakpoints in EWSR1-ATF1 fusions. Detecting all possible gene fusions would require an extensive set of primers, making RT-PCR less feasible in routine diagnostic settings.
- RNA-Based NGS: The Archer FusionPlex assay, an NGS-based anchored multiplex PCR, is a more effective alternative. This method targets a panel of anchored genes, including EWSR1, and sequences across the transcript breakpoint to identify the fusion partner.
Conclusion
AFH remains a diagnostically challenging tumor due to its variable clinical presentation and nonspecific imaging and histological features. However, the integration of immunohistochemistry and molecular techniques has greatly enhanced the ability to accurately diagnose these tumors. The series of patients presented here, along with the review of literature, underscore the importance of wide local excision as the cornerstone of treatment, given its impact on recurrence risk. While AFH generally has a favorable prognosis with a low risk of metastasis, the potential for recurrence necessitates careful long-term follow-up. Continued research and collaboration are essential to further refine diagnostic criteria and optimize treatment strategies for this rare and complex tumor.
References
- Weiss AR, Chen YL, Scharschmidt TJ, et al. Outcomes After Preoperative Chemoradiation With or Without Pazopanib in Non-Rhabdomyosarcoma Soft Tissue Sarcoma: A Report From Children's Oncology Group and NRG Oncology. J Clin Oncol 2023;41(31):4842-4848. DOI: 10.1200/JCO.23.00045.
- Argani P, Harvey I, Nielsen GP, et al. EWSR1/FUS-CREB fusions define a distinctive malignant epithelioid neoplasm with predilection for mesothelial-lined cavities. Mod Pathol 2020;33(11):2233-2243. DOI: 10.1038/s41379-020-0646-5.
To rewatch the webinar, please click the link below. https://forms.gle/esfCgRFreSCae7cX6